Bacterial pathogens are capable of causing illness in people, with infections causing mild/no symptoms to severe illness or even death. Fortunately, we constantly come into contact with many of these bacteria and as such often have a degree of immune system defence against them. Furthermore, the development of new antibiotics over time has meant that most severe bacterial infections people encounter today are treatable, depending on the availability of medication and lack of antibiotic resistance (a topic for another post!). However, occasionally some unlucky person encounters a bacterial pathogen that hasn’t been previously identified, meaning scientists and doctors may be unfamiliar with the illness it can cause, how it causes it, and how best to treat it. Whenever a novel bacterial pathogen is found it needs to be identified before further study can take place.
To identify a novel bacterial pathogen, a sample must first be obtained from the site of infection. If the bacterium is present in an accessible area, such as the upper respiratory tract, ear canal or skin wounds, a swab can be taken. A urine sample may also be sufficient. If the patient is suffering from septicaemia, it may be possible to obtain the bacterium from a blood sample. There may be some of the patient’s native bacterial flora present alongside the pathogen. Hence, it has to be isolated from the other bacteria present in the clinical sample. This can be achieved by streak plating or pour plating if the bacterium is an obligate anaerobe. Once a colony of the pathogen is obtained, bacterial cells can be taken and cultured to produce a genetically homogenous colony of the pathogen for study.
The cells in this homogenous colony are assumed to all have the same genetic material within them. This assumption is only true within the log growth phase of the colony, as bacterial cells have high fidelity DNA replication and basal levels of transcription, meaning the occurrences of genetic mutation are minimised. If the cells are harvested too late, stress on cells will have led to low fidelity replication, transcriptional mutagenesis, and oxidative damage, resulting in a non-homogenous population. This will mean the DNA sample obtained will be of poor quality and limited use. Hence, pathogen cells should be harvested in the early log phase.
The cells are then lysed to release their genetic material. To identify the pathogen, it is best to target genomic DNA; this necessitates gentler methods of cell lysis, to minimise DNA shearing. Gentle lysis is generally achieved through the use of a lysis buffer containing detergents such as SDS/TX-100 (if the bacterium is gram-negative) , buffering salts (e.g. Tris-Cl) and ionic salts (e.g. NaCl), proteases, and TE buffer for DNA solubilisation and protection. Heat may also need to be applied if organic lysis is difficult. Following cell lysis, the lysate is collected. This is an impure sample containing the genomic DNA. To remove remaining protein, lipid and polysaccharide contamination, the lysate is treated with high [salt] and CTAB (a surfactant) and centrifuged. This leads to aggregation of contaminants, which can be removed from the lysate. Some further protein contamination may be found as DNA-bound proteins. These are eliminated through the use of phenol-chloroform extraction. Phenol solubilises and denatures proteins, whilst chloroform allows for fuller separation of the organic and aqueous layer (which contains the genomic DNA), hence preventing DNA degradation. The aqueous layer is collected, and the organic layer discarded. DNA must then be precipitated out of solution through the addition of cold 95%< ethanol/isopropanol and a salt (e.g. sodium acetate). Salt disrupts the water-DNA interactions, making the DNA more hydrophobic, and the addition of alcohol reinforces this effect by ‘shielding’ the DNA-salt complex, reducing the presence of H2O molecules around the DNA. The solution is then centrifuged and the supernatant is removed carefully, to avoiding disturbing the DNA pellet. This pellet is then washed with 70% ethanol several times to remove residual salts. It is then treated with an aqueous solution containing a weak RNase to remove any RNA contamination of the DNA. After washing, the DNA precipitate is resolubilised to obtain a pure DNA sample that can be studied.
Whole genome sequencing (WGS) can now be used to identify the bacterium. As this is a novel bacterium, de novo sequencing methods will be used. First, preparation of a sequencing library is required. Genomic DNA is broken into random fragments; this can be achieved by sonication, hydrodynamic shearing of DNA by soundwaves. The random nature of the DNA fragments is essential to successful composition of an accurate contiguous sequence. These fragments are then ligated to adaptors (short dsDNA molecules) and denatured, producing ssDNA fragments that bind to immobilising elements via the adaptor. The resulting fragments need to be amplified by PCR before sequencing. The adaptors now play a secondary role: serving as universally known short sequences to which the PCR primers can anneal. The fragments are heated in rotation in a solution of free nucleotides and the PCR commences.
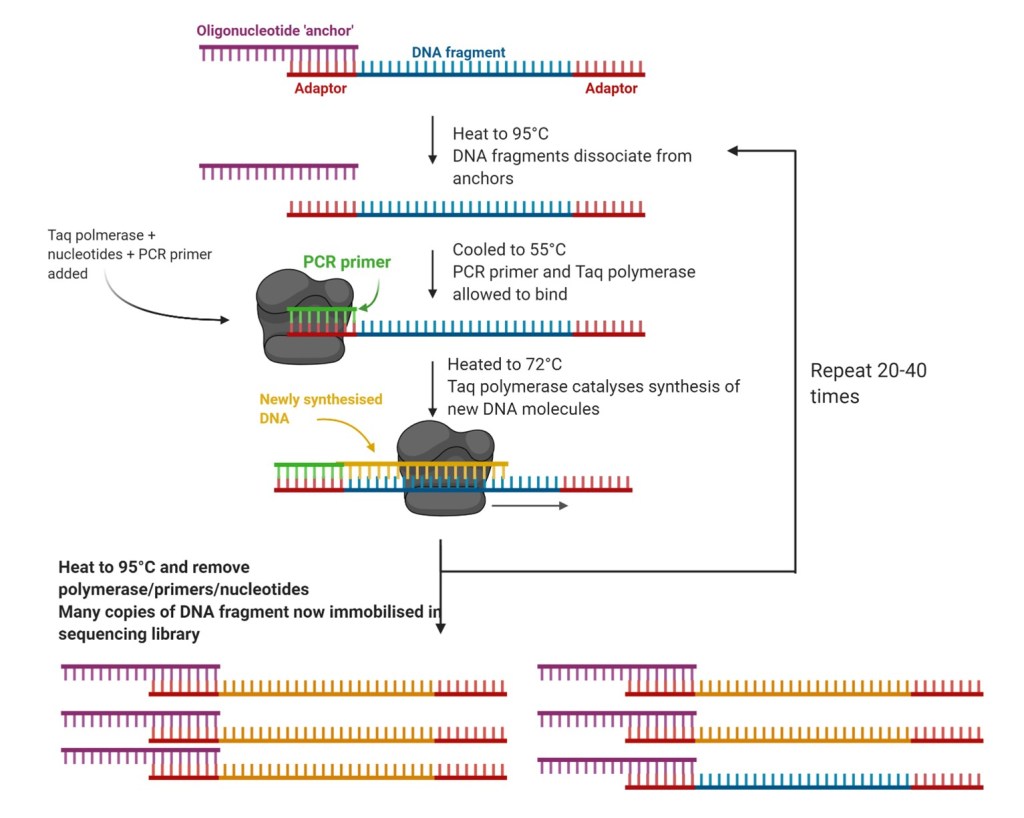
The sequencing library is now prepared. Next-generation sequencing (NGS) methods can be applied to construct a genome sequence. There are multiple NGS methods, but reversible termination sequencing (RTS) is effective for de novo sequencing. RTS involves the use of fluorescently labelled blocker nucleotides that reversibly terminate DNA synthesis following incorporation by polymerase into the DNA molecule. DNA replication is initiated by a primer that binds to the adaptors at the ends of a fragment. A detector identifies the fluorescent tag of the base most recently incorporated, then an enzyme removes the tag, allowing the next base to be incorporated, which is then identified, and so on. This process continues until all the DNA fragments have been sequenced. Due to being run in a massively parallel array, sequencing occurs simultaneously for all fragments.
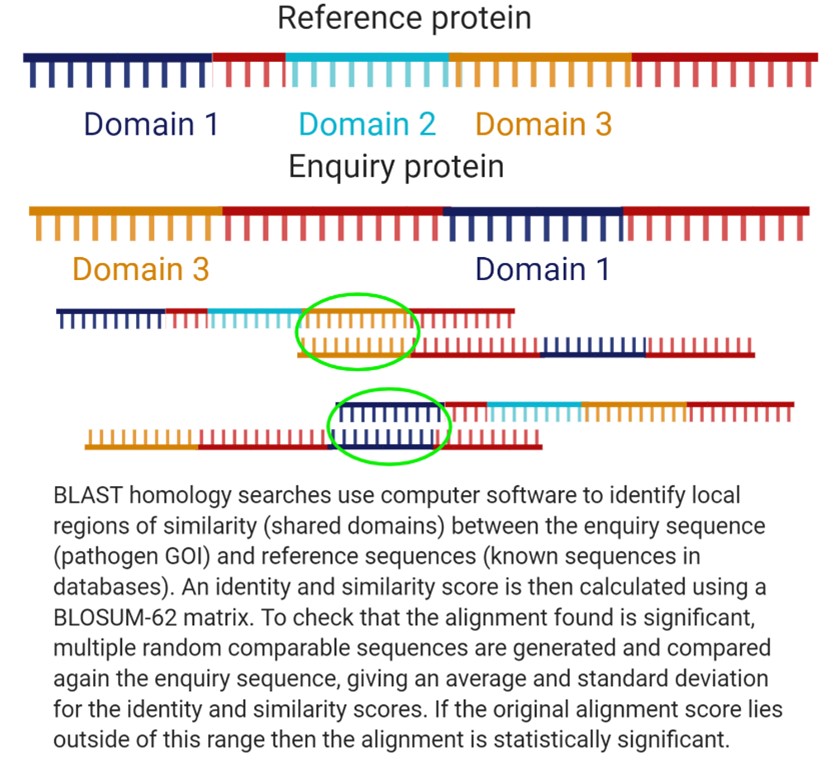
After sequencing all of the genomic DNA fragments, computer software can analyse the fragments and generate a ‘contig read’ by identifying overlapping regions of the DNA fragments. The accuracy of de novo sequencing may be limited by the lack of prior information about the bacterium, so the genome may have to be sequenced multiple times. Once a full genome sequence is obtained, homology searching of databases of known bacterial genomes can be used to identify the pathogen’s family, and potentially regions of interest within the genome. Homology searches are used to identify evolutionarily related genes and rely on computer algorithms such as BLAST to identify homologous sequences. In this case, combining BLAST with other software such as BIGSdb may be effective in identifying the pathogen.
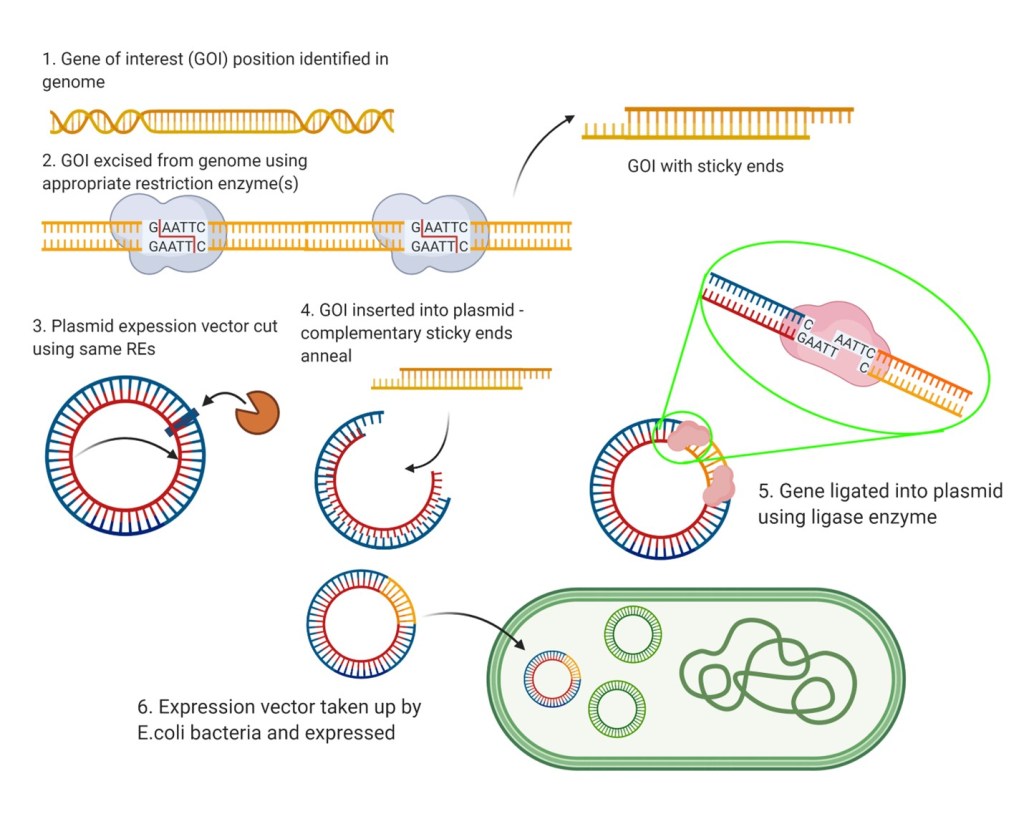
Following homology searching, researchers can isolate and investigate genes of interest (GOIs) by recombinant expression of their proteins for study. GOIs can excised from the bacterium’s DNA by using type II restriction endonucleases (REs). The use of REs means that the resulting gene fragment(s) will have sticky ends, allowing the gene to be incorporated into a plasmid expression vector which can then be incorporated into E. coli cells and expressed. The resulting proteins can then be isolated from the E. coli colony and studied through use of protein assays, X-ray crystallography for structural determination, and more. Studying these proteins may provide clues on the basis of the novel bacterium’s pathogenicity, as well as possible avenues for intelligent drug design. They may also indicate mechanisms of bacterial drug-resistance, crucial for development of effective treatments.